# /// script
# requires-python = ">=3.10"
# dependencies = [
# "annsel",
# "numpy",
# "scanpy",
# ]
# ///
All of Annsel#
This notebook tells you just about everything you need to use annsel. It’s a good starting point to get a feel for the package.
Note
You should be familiar with AnnData beforehand.
Set up Data#
import annsel as an
import numpy as np
import scanpy as sc
We will load a Leukemic bone marrow cytometry dataset :cite:p:Triana2021 You can view the dataset on CellxGene.
adata = an.datasets.leukemic_bone_marrow_dataset()
View the contents of the AnnData object.
display(adata)
AnnData object with n_obs × n_vars = 31586 × 458
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
Importing annsel will automatically register the accessor and add the an attribute to the AnnData object’s namespace.
You can access the methods of the accessor using the an attribute on an AnnData object.
anndata.AnnData.an.<method_name>
Filter#
We can filter the AnnData object using the filter method. Filtering can be applied to the following attributes of an AnnData object:
obsvarvar_namesobs_namesXand its layersobsmandvarmmatrices as a key-value pair containing the attribute’s key name and the predicate to filter on.
Let’s filter the AnnData object by the Cell Type stored in obs["Cell_label"].
adata.an.filter(obs=an.col(["Cell_label"]) == "CD8+CD103+ tissue resident memory T cells")
View of AnnData object with n_obs × n_vars = 890 × 458
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
Note
This is equivalent to
adata[adata.obs["Cell_label"] == "CD8+CD103+ tissue resident memory T cells", :]
You can also apply other expressions to an.col callable, for example is_in allows you to filter by a list of values. Under the hood, annsel uses Narwhals to apply these expressions. A full list of expressions which can be applied to a column can be found here.
adata.an.filter(obs=an.col(["Cell_label"]).is_in(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"]))
View of AnnData object with n_obs × n_vars = 10825 × 458
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
Note
This is equivalent to
adata[adata.obs["Cell_label"].isin(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"]), :]
We can also combine multiple Predicates using the & and | operators.
adata.an.filter(
obs=(
an.col(["Cell_label"]).is_in(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"]),
an.col(["sex"]) == "male",
),
var=an.col(["vst.mean"]) >= 3,
obsm={"X_pca": an.col([0]) > 0},
)
View of AnnData object with n_obs × n_vars = 736 × 67
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
adata.an.filter(
obs=(an.col(["Cell_label"]).is_in(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"]))
& (an.col(["sex"]) == "male")
)
View of AnnData object with n_obs × n_vars = 7727 × 458
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
Note
This is equivalent to
adata[
(adata.obs["Cell_label"] == "CD8+CD103+ tissue resident memory T cells")
& (adata.obs["sex"] == "male")
]
Or if you pass a tuple of Predicates, it will apply the & operator between them automatically.
adata.an.filter(
obs=(
an.col(["Cell_label"]).is_in(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"]),
an.col(["sex"]) == "male",
)
)
View of AnnData object with n_obs × n_vars = 7727 × 458
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
Note
This is equivalent to
adata[
(adata.obs["Cell_label"].isin(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"]))
& (adata.obs["sex"] == "male")
]
The | operator is applied between the Predicates as well. Here we will select all the cells that are either Classical Monocytes or CD8+CD103+ tissue resident memory T cells, or are from male samples irrespective of the cell type.
adata.an.filter(
obs=(
an.col(["Cell_label"]).is_in(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"])
| (an.col(["sex"]) == "male")
)
)
View of AnnData object with n_obs × n_vars = 18868 × 458
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
Note
This is equivalent to
adata[
(adata.obs["Cell_label"].isin(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"]))
| (adata.obs["sex"] == "male")
]
We can also filter the AnnData object by the var column. Here we will filter the AnnData object to only include the genes with vst.mean greater than or equal to 3.
adata.an.filter(var=an.col(["vst.mean"]) >= 3)
View of AnnData object with n_obs × n_vars = 31586 × 67
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
Note
This is equivalent to
adata[adata.var["vst.mean"] >= 3]
Filtering can also be applied to the X matrix. Here we will filter the AnnData object to only include the cells with ENSG00000205336 gene expression greater than 1. If you want to filter by a layer, you can pass the layer name to the layer argument. and the operation will be applied to the var_name of that layer.
This dataset does not include a layer, so let’s create our own layer. We will arcsinh transform the X matrix and store it in a new layer called arcsinh.
adata.layers["arcsinh"] = np.arcsinh(adata.X)
adata.an.filter(
x=an.col(["ENSG00000205336"]) > 1,
layer="arcsinh",
)
View of AnnData object with n_obs × n_vars = 6709 × 458
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
layers: 'arcsinh'
This can all be combined together as well
adata.an.filter(
obs=(
an.col(["Cell_label"]).is_in(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"]),
an.col(["sex"]) == "male",
),
var=an.col(["vst.mean"]) >= 3,
)
View of AnnData object with n_obs × n_vars = 7727 × 67
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
layers: 'arcsinh'
Note
This is equivalent to
adata[
(adata.obs["Cell_label"].isin(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"]))
& (adata.obs["sex"] == "male"),
(adata.var["vst.mean"] >= 3),
]
Filtering can also be applied to var_names and obs_names using the an.var_names and an.obs_names predicates respectively. These are special predicates which can only be applied to var_names and obs_names of the AnnData object.
an.obs_names = an.col("obs_names") and an.var_names = an.col("var_names"). They are simply special instances of an.col.
Here, arbitrarily, we will filter the AnnData object to only include the cells with obs_names starting with 645 and the genes with var_names starting with ENSG0000018.
adata.an.filter(obs_names=an.obs_names.str.starts_with("645"), var_names=an.var_names.str.starts_with("ENSG0000018"))
View of AnnData object with n_obs × n_vars = 37 × 30
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
layers: 'arcsinh'
For obsm and varm matrices, filtering is a bit less intuitive, since those attributes are less restricted on their underlying data type (NumPy arrays, Xarray, Pandas, etc…) your milage may vary. In addition the column names are numerical indices starting from 0.
Here we will filter the obsm matrix to only include the cells with X_pca greater than 0.
For this example dataset the X_pca matrix is a NumPy array.
adata.an.filter(obsm={"X_pca": an.col([0]) > 0})
View of AnnData object with n_obs × n_vars = 17005 × 458
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
layers: 'arcsinh'
Select#
We can also apply the select method to the AnnData object. This is similar to the filter method, but it will only keep the rows and columns that match the Predicates. It can be applied to the obs, var, and X.
Here we will select the Cell_label and sex columns from the obs table, the feature_name column from the var table and the ENSG00000205336 gene from X. This will return a new AnnData object with only these columns in the obs, var and X tables.
adata.an.select(
obs=an.col(["Cell_label", "sex"]),
var=an.col(["feature_name"]),
x=an.col(["ENSG00000205336"]),
)
AnnData object with n_obs × n_vars = 31586 × 1
obs: 'Cell_label', 'sex'
var: 'feature_name'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
layers: 'arcsinh'
Group By#
Group By’s can be performed on both the obs and var DataFrames, even at the same time. They will return a generator consisting of GroupData objects, which contain the grouped AnnData object, the names of the obs and var columns used to group by as well as the values for those columns in that group.
For example if we group by the Cell_label column in the obs table and the feature_type column in the var table, we will get a generator of GroupData objects.
And that first element will look like the following:
first_group = next(adata.an.group_by(
obs=an.col(["Cell_label"]),
var=an.col(["feature_type"]),
copy=False
)
Note that if copy=False, the GroupData object will be a view of the original AnnData object. If you want a copy, you can set copy=True.
GroupByAnnData:
├── Observations:
│ └── Cell_label: Lymphomyeloid prog
├── Variables:
│ └── feature_type: protein_coding
└── AnnData:
View of AnnData object with n_obs × n_vars = 913 × 445
...
For a given group object you can access the following attributes:
obs_dict: A dictionary of the groupby column and the value for that obs group.{'Cell_label': 'Lymphomyeloid prog', 'donor_id': 'AML1'}
var_dict: A dictionary of the groupby column and the value for that var group.{'feature_type': 'protein_coding'}
obs_values: A tuple of the values of the groupby column for that obs group.('Lymphomyeloid prog', 'AML1')
var_values: A tuple of the values of the groupby column for that var group.('protein_coding',)
first_group = next(
adata.an.group_by(
obs=an.col(["Cell_label", "donor_id"]),
var=an.col(["feature_type"]),
)
)
print(f"obs_values: {first_group.obs_values}")
print(f"var_values: {first_group.var_values}")
print(f"obs_dict: {first_group.obs_dict}")
print(f"var_dict: {first_group.var_dict}")
obs_values: ('Lymphomyeloid prog', 'AML1')
var_values: ('protein_coding',)
obs_dict: {'Cell_label': 'Lymphomyeloid prog', 'donor_id': 'AML1'}
var_dict: {'feature_type': 'protein_coding'}
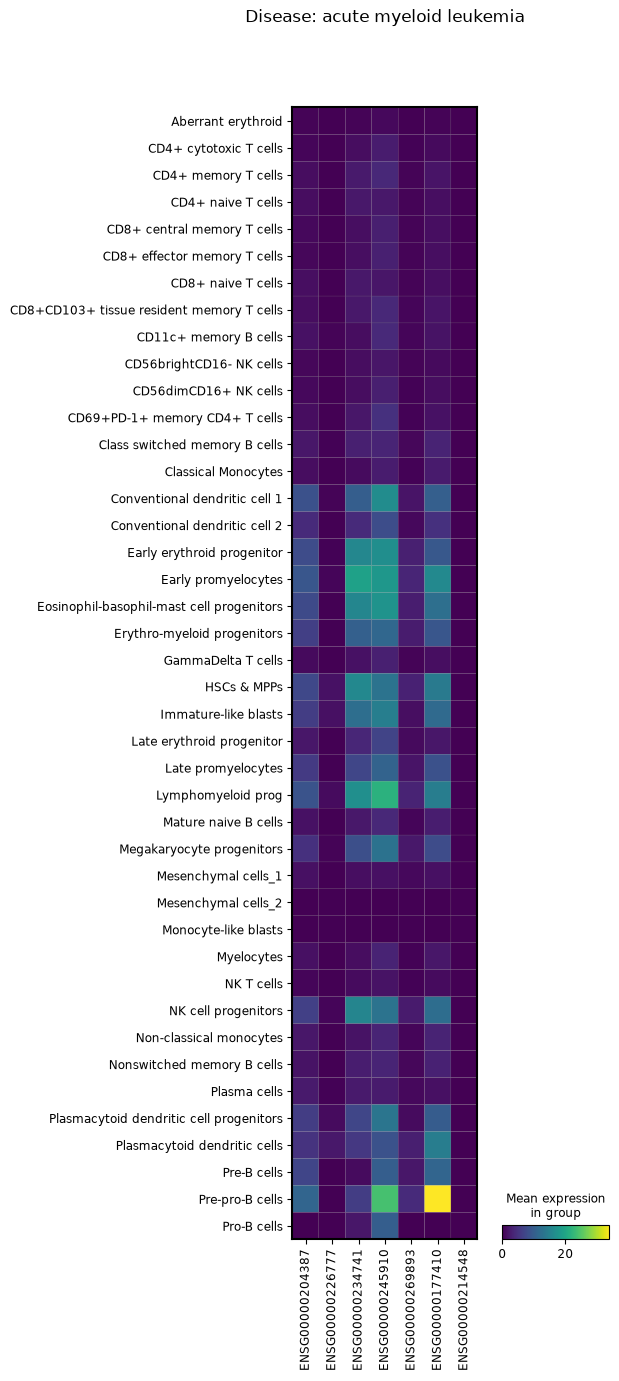
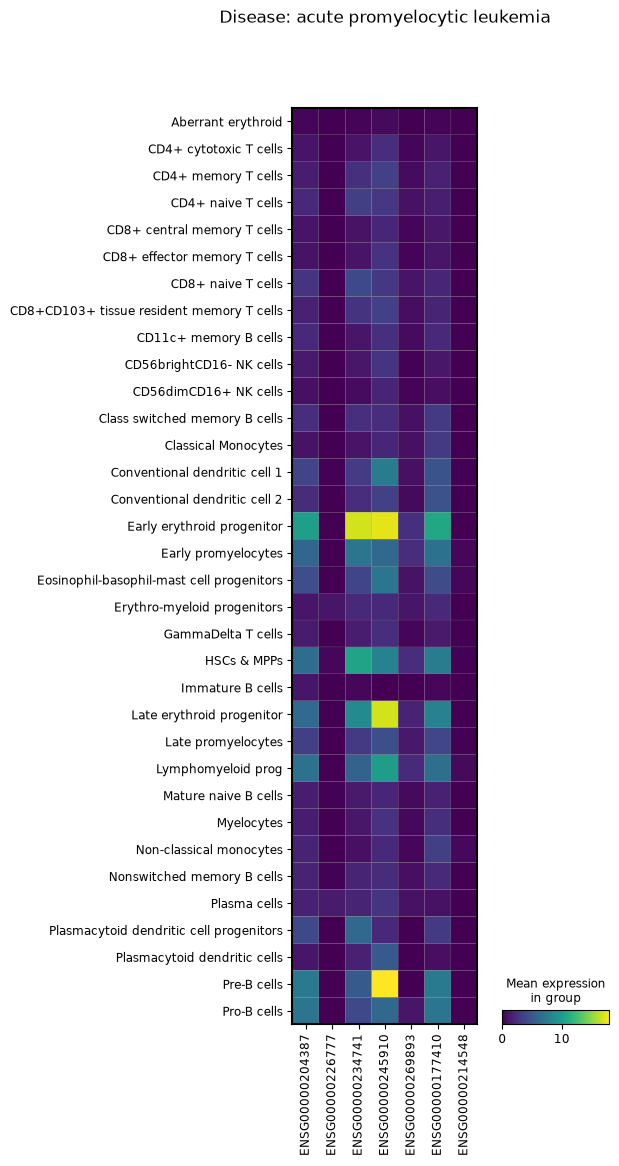
This example demonstrates how to compare gene expression patterns across different disease conditions by creating separate visualizations for each disease group.
We’ll group the AnnData object by disease, filter for specific gene types (lncRNAs), and generate matrix plots showing expression patterns across cell types for each condition.
adata.an.group_by(
obs=an.col(["Cell_label"]),
copy=False,
)
<generator object _group_by at 0x7ae061771640>
next(
adata.an.group_by(
obs=an.col(["Cell_label"]),
copy=False,
)
)
GroupByAnnData:
├── Observations:
│ └── Cell_label: Lymphomyeloid prog
├── Variables:
│ └── (all variables)
└── AnnData:
View of AnnData object with n_obs × n_vars = 913 × 458
obs: 'Cluster_ID', 'donor_id', 'Sample_Tag', 'Cell_label', 'is_primary_data', 'organism_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'assay_ontology_term_id', 'tissue_ontology_term_id', 'Genotype', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'disease_ontology_term_id', 'cell_type_ontology_term_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable', 'feature_is_filtered', 'Unnamed: 0', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'cell_type_ontology_term_id_colors', 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_bothumap', 'X_pca', 'X_projected', 'X_projectedmean', 'X_tsneni', 'X_umapni'
layers: 'arcsinh'
for group in adata.an.group_by(obs=an.col(["disease"])):
_adata = group.adata.an.filter(var=(an.col(["feature_type"]) == "lncRNA"))
_plot = sc.pl.matrixplot(
_adata,
var_names=_adata.var_names,
groupby="Cell_label",
return_fig=True,
title=f"Disease: {group.obs_dict['disease']}",
)
_plot.show()
Pipe#
We can also use pipe to apply functions on AnnData objects.
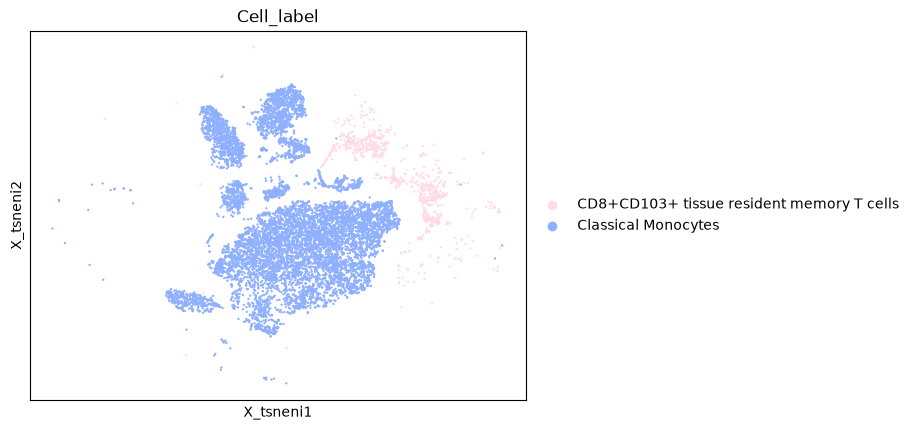
adata.an.pipe(sc.pl.embedding, basis="X_tsneni", color="Cell_label")

We can chain together multiple methods as well.
adata.an.select(obs=an.col(["Cell_label"])).an.filter(
obs=an.col(["Cell_label"]).is_in(["Classical Monocytes", "CD8+CD103+ tissue resident memory T cells"])
).an.pipe(sc.pl.embedding, basis="X_tsneni", color="Cell_label")